
药物分析
2022-03-15 23:17:08 6 举报AI智能生成
关于药物的杂质相关知识,包括杂质的检查等
药学
药物分析
模板推荐
作者其他创作
大纲/内容
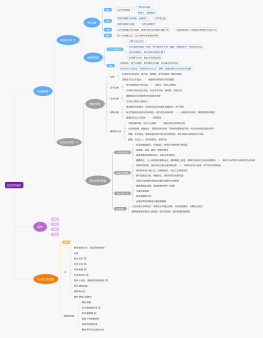
第二章 药物的鉴别实验
药物的鉴别方法
化学鉴别法
呈色反应鉴别法
沉淀生成反应鉴别法
荧光反应鉴别法
药物在适当溶剂中在可见光下发射荧光//药物与试剂反应发射荧光
气体生成鉴别法
使试剂褪色的鉴别法
制备衍生物测定熔点鉴别法
光谱鉴别法
紫外光谱鉴别法
最大吸收波长
最小吸收波长
吸收度
吸收波长与吸收度的比值法
比较光谱图
肩峰,峰值
红外光谱鉴别法IR
标准图谱对照法
近红外光谱法
原子吸收法
核磁共振法
质谱鉴别法
粉末X射线衍射法
色谱鉴别法
薄层色谱鉴别法
斑点一致
高效液相色谱和气相色谱鉴别法
保留时间一致
显微鉴别法
中药及其制剂的鉴别
生物学法
抗生素,生化药物,中药的鉴别
指纹图谱与特征图谱鉴别法
中药
第三章
第一节药物的杂质与限量
药物的杂质与纯度
杂质
任何<b><font color="#d32f2f">影响药品纯度</font></b>的物质均称为杂质。(无治疗作用,或影响药物的稳定性和疗效,甚至对人体健康有害。)
纯度
指药物的纯净程度,是反映药品质量的一项重要指标
主要由药品质量标准中“检查”项下的杂质检查来控制
药物纯度的评价
应把药物的性状,理化常数,杂志检查,含量测定等作为一个有联系的整体来评价药物的纯度
药物纯和化学纯
化学纯度
不考虑杂质的生理作用,其限量只是从可能引起的化学变化对使用的影响来限定
药物纯度
主要从用药安全,有效和对药物稳定性等方面来考虑
药物杂质的来源
生产过程中引入
药物在合成过程中<font color="#f44336">未反应的原料,中间体和副产物</font>,所用的<font color="#e57373">试剂</font>,<font color="#e57373">溶剂</font>,<font color="#f44336">还原剂,金属器皿装置</font>等
原料药生产中引入的杂质
制剂生产中引入的杂质
无效,低效异构体或晶型(光学异构体,几何异构体,多晶型)
贮藏过程中引入
<font color="#f44336">外界条件</font>的变化或<font color="#f44336">微生物</font>的作用可能发生的水解,氧化,异构化,晶型转化,聚合等变化而产生的有关杂质
易发生水解反应(酯,内酯,酰胺,卤代烃,苷类)
易发生氧化反应的结构(醛,醚,酚羟基等)
严格控制药品的贮藏条件,是保证临床用药安全,有效的一个重要方面
药物杂质的分类
按来源分
一般杂质
在自然界中分布比骄傲广泛,在多种药物的生产和贮藏过程中容易引入的杂质,如酸,碱,水分,氯化物,硫酸盐,重金属,砷盐等
特殊杂质
在特定药物的生产和贮藏过程中引入的杂质,如阿司匹林中游离水杨酸等
按毒性分
信号杂质
本身一般无毒,但其含量的多少可以反映出药物的纯度水平,称其为“信号杂质”,如氯化物,硫酸盐等属于信号杂质
毒性杂质
对人体有毒害的杂质。如重金属,砷盐,氯化物等应严格控制,以保证用药安全
按化学类别和特性分类
无机杂质
来源于生产过程中,一般是已知的和确定的,直接影响药物的稳定性,反映生产工艺情况
有机杂质
合成中未反应完全的原料,中间体,副产物,降解产物等
残留溶剂
生产中使用的有机溶剂
杂质的限量
定义
药物中所含杂质的最大允许量
表示
百分之几或百万分之几ppm
计算
杂志限量=杂质标准溶液(浓度*体积)/供试品量*100%
L=c*V/S*100%
检查方法
限度检查法
对照法
系指取一定量的被检杂质标准溶液和一定量供试品溶液,在相同条件下处理,比较反应结果,以确定杂质含量是否超过限量。
注意平行原则
灵敏度法
系指在供试品溶液中加入一定量的试剂,在一定反应条件下,不得有正反应出现,从而判断供试品中所含杂质是否符合限量规定
比较法
系指取供试品一定量依法检查,测定特定待检杂质的参数(吸光度等)与规定的限量比较,不得更大
定量测定
第二节 杂志的检查方法
杂志的常用检查方法
化学方法
显色反应检查法
沉淀反应检查法
生成气体检查法
滴定法
色谱方法
色谱法是有关物质检查的首选方法
薄层色谱法
杂质对照品法
供试品溶液的自身稀释对照法
两法并用法
对照药物法
高效液相色谱法
外标法
加校正因子的主成分自身对照法
不加校正因子的主成分自身对照法
面积归一化法
气相色谱法
同上
标准溶液加入法
毛细管电泳法
多肽,酶类
光谱方法
紫外可见分光光度法
红外分光光度法
原子吸收分光光度法
其他方法
热分析法
酸碱度检查法
物理性状检查法
第三节 药物中一般杂质的检查
氯化物检查法
原理
氯化物在硝酸条件下与硝酸银反应生成氯化银白色沉淀,与标准溶氯化钠对比,判定限量。
方法
取供试品加水溶解成约25ml,加<font color="#f44336">稀硝酸</font>10ml,置于50ml纳氏比色管,加水摇匀即得<b>供试品溶液</b>;取规定的标准氯化钠溶液,置于50ml纳氏比色管,加稀硝酸10ml,加水即为<b>对照溶液</b>。分别加入硝酸银溶液1.0ml用水稀释至50ml。在暗处放置5min,<b>同置于黑色背景上,从比色管上方向下观察</b>,比较,即得。
注意事项
根据标准氯化钠中氯化物的浓度,待测氯化物浓度以<b>50ml中含50-80ugCl</b>为适宜。
加<b>硝酸</b>可以避免如<font color="#f44336">碳酸根,磷酸根</font>等弱酸根盐的干扰,并且可以加速氯化银沉淀。以50ml供试品含10ml为适宜
滤液不澄清用不含氯化物的<b>滤纸过滤</b>。
供试品带有颜色,用<b>内消色法</b>消除干扰
内消色法
取两分供试品溶液,一份加入硝酸银溶液,反复滤过沉淀直至溶液澄清,<br>再加入标准氯化钠溶液与水适量制成对照溶液,剩余方法与上述一致。
硫酸盐检查法
原理
硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成白色硫酸钡沉淀,在和标准硫酸钾进行对比,判定限量
方法
取供试品,加入溶解成40ml,置于50m纳氏比色管,加2ml<b>稀盐酸</b>,即得<font color="#f44336">供试品溶液</font>;另取标准硫酸钾溶液,置于纳氏比色管,加水至40ml,即得<font color="#f44336">对照溶液</font>。分别加入25%氯化钡5ml,稀释至50ml。放置10min,<b>同置于黑色背景板上,从比色管上方向下观察</b>,比较,即得。
注意事项
硫酸钾100ugSO4/ml
<b>盐酸</b>的作用是防止<font color="#f44336">碳酸钡或磷酸钡</font>等弱酸形成钡盐沉淀干扰,但是<font color="#f44336">酸度过大</font>会使硫酸钡溶解,<font color="#f44336">灵敏度降低</font>。以50ml含2ml为适宜
供试品如果带有颜色,可以采用<b>内消色法</b>消除
内消色法
取供试品两份,一份加入规定氯化钡溶液,反复过滤直到溶液澄清,<br>加入规定量硫酸钾溶液,作为对照溶液,供试品溶液制备方法与上述相同。
如果药物在<b>水中不易溶解</b>,可加入适量的与水互溶的<font color="#f44336">有机溶剂</font>将药物溶解,使被包裹的待检查杂质释放后,再依法检查。
铁盐检查法
铁盐可以加速药物的氧化和降解
原理
铁在盐酸酸性条件下与硫氰酸盐作用生成<b>红色可溶</b>性的硫氰酸铁配离子,与标准铁溶液对比判定。
方法
取适量供试品,加水溶解至25ml,置于纳氏比色管,加<font color="#f44336">稀盐酸</font>4ml与<font color="#f44336">过硫酸铵</font>50mg,用水稀释至35ml,加30%<font color="#f44336">硫氰酸铵</font>3ml,加水稀释至50ml,如果显色,立即制备对照溶液。取标准铁溶液,置于50ml纳氏比色管,加水至25ml,加稀盐酸4ml和过硫酸铵50mg,用水稀释至35ml,加30%硫氰酸铵3ml,加水稀释至50ml,比较。
注意事项
标准铁溶液--10ug/ml。硫酸铁铵配置,加<b>硫酸</b>防止<b>铁盐水解</b>
称取硫酸铁铵0.863g----1000ml量瓶,作贮备液。临用先取
当50ml溶液中含5-90ug时,溶液吸光度与浓度呈良好线性关系。目视比色效果适宜。
<b>盐酸酸性</b>条件下反应,可以<font color="#f44336">防止</font>三价铁离子<font color="#f44336">水解</font>。50ml含稀盐酸4ml为宜
加入<b>过硫酸铵</b>氧化剂,既可以<b>氧化供试品</b>中的二价铁为三价铁,还可以<font color="#f44336">防止</font>由于<font color="#f44336">光线</font>使得硫氰酸铁还原或分解<font color="#f44336">褪色</font>
某些药物如葡萄糖,糊精,和硫酸镁等,在检查过程中需要加入<b>硝酸</b>处理,硝酸也可将二价铁离子<font color="#f44336">氧化</font>成三价铁离子。因为硝酸中可能含有亚硝酸,它能与硫酸根离子作用,生成红色亚硝酸硫氰化物,影响比色,所<font color="#f44336">以剩余的硝酸需要加热煮沸</font><b>除去</b>
铁盐与硫氰酸根离子反应是<b>可逆反应</b>,加入过量的<b>硫氰酸铵</b>,不仅可以<font color="#e57373">增加</font>生成的 配位离子的<font color="#f44336">稳定性</font>,提高反应<font color="#e57373">灵敏度</font>,还能<font color="#e57373">消除</font>因其他阴离子以铁盐形成配位化合物而引起的<font color="#e57373">干扰</font>
若供试品溶液管与对照液管<b>色调不一致</b>,或所呈现硫氰酸铁的颜色较浅不易比较时,可分别转移至分液漏斗中,各加入<b>正丁醇</b>或<b>异丁醇</b>20ml<b>提取</b>,待分层后,将正丁醇层移至50ml纳氏比色管中,再用正丁醇稀释至25ml,比较,即得。<br>硫氰酸铁配位离子在正丁醇等有机溶剂中的<b>溶解度较大</b>,上述处理能增加颜色深度,同时可以排出上述酸根离子的影响。
某些有机药物特别是<b>具有环状结构</b>的有机 药物,在试验条件下不溶解或对检查有干扰,则需要经过<b>炽灼破坏</b>,使铁盐转变为三氧化二铁留于残渣中,处理后在依法检查。
重金属检查法
药品生产中遇到铅的机会较多,且铅容易蓄积中毒,故均已铅为重金属的代表,以<b>铅 的限量表示重金属限度</b>
第一法 硫代乙酰胺法
适用
溶于水,稀酸或与水互溶有机溶剂,不含与金属离子强配位基团的药物
原理
硫代乙酰胺在弱酸性条件下水解,产生硫化氢,可以与重金属离子生成黄色到棕黑色的硫化物混悬液。
方法
25ml纳氏比色管3支,<b>甲管-标准管</b>--加标准铅溶液加醋酸盐缓冲液;<b>乙管-供试品管</b>-供试品溶液;<b>丙管-标准加样管</b>-供试品加标准铅溶液和醋酸盐缓冲液。分别加硫代乙酰胺。<b>同置白纸上,自上而下透视,丙管>甲管>乙管</b>
注意事项
标准铅溶液--加<b>硝酸</b>防止<font color="#f44336">铅盐水解</font>。
供试品有<b>颜色</b>,<font color="#e57373">甲</font>管加入少量稀焦糖溶液或其他无干扰的<font color="#f44336">有色溶液</font>,颜色一致,加入硫代乙酰胺比较。如果无法比较,用第二法
供试品含有<b>高铁盐</b>,弱酸条件容易氧化硫化氢析出硫。分别加入<b>维生素C</b>,<font color="#f44336">还原高铁离子为亚铁离子</font>。
金属离子与硫化氢的呈色,受<b>pH</b>影响大,3-3.5--沉淀完全,酸度增大,呈色浅,甚至不显色。因此如果供试品用强酸溶解,要用氨水调至中性后,在加入pH3.5的醋酸盐缓冲液调节酸度
配置供试液时,用盐酸超过1ml,氨试液超过2ml或加入其他试剂进行处理者,为避免标准管的基质差异,应当进行<b>平行处理</b>。除另有规定外,甲管应取同样试剂在<font color="#f44336">瓷皿中蒸干</font>,加醋酸盐缓冲液和水,微热溶解后,移至比色管,加标准量铅溶液,加水稀释
第二法 炽灼后的硫代乙酰胺法
适用
难溶于水,稀酸,与水互溶有机溶剂的有机药物,含可与金属离子强配位基团的芳环和杂环药物
原理
重金属会与含有强配位基团的药物形成价键作用;或者供试品不溶解,可能包裹重金属。要先将其炽灼破坏,加硝酸进一步破坏,蒸干。加盐酸转化为易溶水的氯化物,再按第一法检查
方法
供试品<b>按炽灼残渣法炽灼</b>,再取遗留的残渣,或直接取炽灼残渣项下遗留的残渣,加硝酸,蒸干,至氧化氮蒸气除尽后,放冷,加盐酸,蒸干加水,加氨试液,调节pH,在加醋酸缓冲液,微热溶解,置于纳氏比色管,加水稀释,作为乙管--供试品管。 另取供试品溶液,置于瓷皿中蒸干加入醋酸盐缓冲液和水,微热溶解,置于比色管,加入标准铅溶液,作为甲管--标准管。 <b>甲乙</b>分别加入硫代乙酰胺,<b>同置于白纸上,自上而下透视</b>,甲管颜色>乙管
炽灼残渣处理过程中,温度越高,重金属损失越多。因此,炽灼温度对重金属的检查结果影响较大。应该控制温度在500-600℃,同时要控制时间
注意事项
炽灼残渣加硝酸加热,必须<b>蒸干</b>,除尽氧化氮,否则<font color="#f44336">亚硝酸可氧化硫化氢析出硫</font>,影响比色
为了消除盐酸或者其他试剂中夹杂<font color="#f44336">重金属</font>的影响,在配置供试品溶液时,使用盐酸超过1ml,使用氨溶液超过2ml,以及用硫酸与硝酸进行有机破坏或其他试剂处理者,除另有规定外,甲管应取同样同量试剂置瓷皿中蒸干后,依法检查
含<b>钠盐和氟</b>的有机药物,在炽灼时能腐蚀瓷坩埚,而引入重金属,应该改用<b>铂坩埚</b>或更<b>硬质玻璃器蒸发皿</b>
如乳酸钠
第三法 硫化钠法
适用
溶于碱性水溶液而难溶于稀酸或在稀酸中即生成沉淀的药物---磺胺类,巴比妥类
原理
硫化钠与铅生成硫化铅
方法
取供试品适量,加入氢氧化钠和水溶解,置于纳氏比色管,加入硫化钠,与标准铅溶液同法处理后比较颜色
注意事项
硫化钠对玻璃有一定腐蚀性,久置产生絮状物,要<b>临用新制</b>
饱和硫化氢水溶液:上述方法中使用的硫化钠或硫代乙酰胺,均可以使用<b>新制的饱和硫化氢</b>水溶液替代。
硫化氢制备--硫化铁与稀盐酸作用,导气管引入纯净水中被吸收,即得饱和硫化氢溶液
砷盐检查法
砷盐多由药物生产过程中使用的无机试剂引入
第一法 古蔡氏法
原理
锌与酸作用产生新生态的氢,与砷盐反应,生成具有挥发性的砷化氢,遇到溴化汞试纸,产生黄色到棕色砷斑。与标准珅溶液的砷斑对比判定
方法
仪器装置
见图1
标准砷斑的制备
标准砷溶液,加盐酸,水,加碘化钾与酸性氯化亚锡,5min后加锌,立即安装导气管,水浴,45分钟后检查砷斑
检查法--样品砷斑的制备
取供试品溶液,加碘化钾与酸性氯化亚锡,5min加锌,立即安装导气管,水浴,45min后检查砷斑
注意事项
标准砷溶液的制备---用<font color="#e57373">三氧化二砷</font>配置<b>贮备液</b>,临用前用稀硫酸定量稀释配置
碘化钾和氯化亚锡
碘化钾及氯化亚锡的催化作用----碘化钾和氯化亚锡可以<b>还原</b><font color="#e57373">五价</font>砷为<font color="#e57373">三价</font>砷,碘化钾被<b>氧化</b>生成的<font color="#e57373">碘</font>,又可以被氯化亚锡还原为<font color="#e57373">碘离子</font>,后者和溶液中的<font color="#e57373">锌</font>离子反应形成稳定的<b>配位离子</b>,有利于生成砷化氢的<b>反应不断进行</b><br>氯化亚锡还可以和锌作用,在其表面形成<b>锌锡齐</b>,起<b>去极化作用</b>,使<font color="#e57373">氢气均匀连续发生</font>。<br>氯化亚锡和碘化钾还可以<b>抑制锑化氢</b>的生成,<font color="#f44336">消除锑化氢</font>对实验<font color="#f44336">的干扰</font>
醋酸铅棉花的作用
锌和供试品中的硫化物产生的硫化氢会干扰实验<b>。醋酸铅棉花</b>可以<b>吸收硫化氢</b>,消除硫化物的干扰。还可以使砷化氢以<font color="#f44336">适宜的速度通过</font>。
仪器和试剂要求
仪器和试液<font color="#f44336">不得生成砷斑</font>。
标准砷斑和供试品砷斑<font color="#f44336">同时进行制备</font>
锌中<font color="#f44336">无砷</font>
溴化汞试纸的制备---滤纸条浸入乙醇制溴化汞试液,1h取出,干燥暗处
溴化汞试纸比氯化汞灵敏,但砷斑不稳定,要干燥避光
含硫药物的预处理
供试品中有含硫化物,在酸性溶液中生成硫化氢或二硫化物气体,和溴化汞试纸作用生成黑色硫化汞或金属汞,产生干扰。供试品预先<b>加硝酸湿法</b>消化处理,使<font color="#f44336">硫化物氧化为硫酸盐</font>,消除干扰
杂环有机药物的预处理
<b>环状结构有机药物</b>,因为砷可能和分子以<font color="#f44336">共价键结合</font>,要预先进行有机<b>破环</b>,常用方法,<font color="#f44336">碱破坏法,酸破坏法</font>
第二法 二乙基二硫代氨基甲酸银法--Ag-DDC
原理
锌与酸作用产生新生态的氢,与砷生成挥发性的砷化氢,可以<font color="#f44336">还原二乙基二硫代氨基甲酸根</font>,产生<b>红色胶态银</b>,用目<font color="#b71c1c">比视法或测定吸光度</font>。检查杂质限量或含量测定。
方法
DDC-Ag检砷装置如图2
标准砷溶液的制备:标准砷溶液加盐酸和水,再加碘化钾和酸性氯化亚锡,再加锌,立即安装导气管,水浴反应,添加三氯甲烷
检查法--供试品溶液,加碘化钾和酸性氯化亚锡,再加锌,同法操作。<b>置于白色背景上,自上而下观察比较</b>。或在510nm的分光光度法测定吸光度。
注意事项
除与第一法相同外,还有
灵敏度范围--AS在<font color="#f44336">1~10ug</font>,所得胶体银溶液稳定,重现性好,而古蔡氏法通常仅限于在<font color="#e57373">2ug</font>的As检查
二乙基二硫代氨基甲酸银溶液--取DDC-Ag0.25g,加三氯甲烷和三乙胺,用脱脂棉滤过,制备。置于棕色瓶内,密封,阴凉处
也可以用<b>吡啶作</b>为吸收反应中产生二乙基二硫代氨基甲酸的试剂。但是吡啶有恶臭,用三乙胺和三氯甲烷溶液,灵敏度略低于吡啶溶液
锑化物的消除--加入酸性<b>氯化亚锡和碘化钾</b>试液,可进一步<font color="#f44336">抑制锑化氢</font>的形成
其他方法
白田道夫法
适用对象
含锑的药物--葡萄糖锑钠
原理
锑化氢干扰试验
<b>氯化亚锡</b>在盐酸中将砷盐还原为<b>棕褐色的胶态砷</b>,于标准砷溶液用同法处理比较,判定
少量氯化汞可以提高灵敏度
次磷酸法
原理
盐酸酸性溶液中,次磷酸将砷盐还原为<b>棕色的游离砷,</b>与标准砷溶液处理后比较,判定
适用对象
硫化物-亚硫酸盐以及含锑药物的砷盐检查,灵敏度比古蔡氏法低
干燥失重测定法
适用对象
水分,其他挥发性物质
常压恒温干燥法
适用对象
受热比较稳定的药物
方法
取供试品,混匀,取一定重量,置于干燥至恒重的扁形称量瓶中,称定,105℃干燥至恒重,计算干燥失重
干燥失重=减失的重量/取样量*100%
注意事项
操作要求,干燥时平铺供试品在容器中,规定厚度,瓶盖放置好。取出时盖好盖子,置于干燥器中放冷,称定。
渐次升高温度干燥法:
某些药物中含有较大量的水分,或者熔点较低,如果直接在105℃直接干燥,供试品容易融化,在表面结成一层薄膜,使水分不易继续挥发。供试品如果未达成规定的干燥温度即融化时,应先将供试品在低熔化温度5-10℃下干燥至大部分水分除去后,在按规定条件干燥。
高温干燥法
含有较多结晶水的药物,在105℃不易除去结晶水,或结晶与吸附溶剂不易失去时,可提高干燥温度。
定时失重法
某些易吸湿或受热发生相变而达不到恒重的药物,采用一定温度下,干燥一定时间所减失的重量代表干燥失重
减压干燥法与恒温减压干燥法
适用对象
熔点低,受热易分解的供试品
原理
采用减压干燥器或恒温减压干燥器。常用干燥剂为五氧化二磷,硫酸,硅胶。
干燥剂干燥法
适用对象
受热分解或易升华的供试品
常用干燥剂--硅胶,硫酸,五氧化二磷。硅胶的吸水能力仅次于五氧化二磷
方法
将供试品置干燥器中,利用干燥器内的干燥剂吸收水分,干燥至恒重
使用<b>五氧化二磷</b>,需要将干燥剂平铺在培养皿中,置于干燥器中。干燥剂表面结块,应该将表面刮去,另外加五氧化二磷再使用。使用后的五氧化二磷要进行无害化处理。
注意事项
使用<b>硫酸</b>时,将硫酸盛于培养皿中或烧杯中,不能直接倾于干燥器中,可以反复使用--将含水硫酸置于烧杯中,加热至冒白烟,保持110℃30min左右。
硅胶为<b>变色硅胶</b>,里面添加有氯化钴。无水氯化钴为蓝色,吸水由蓝色变为蓝紫,紫红转变为粉红色显示硅胶失效。105℃干燥后可以反复使用
水分测定法
适用对象
水分(结合水,吸附水)
费休氏法
原理
碘氧化二氧化硫为三氧化硫时,需要一定的水分参与反应,根据定量反应关系和消耗碘的重量,计算参与反应的水分含量。为了使反应往正反应方向进行,费休氏试液大都使用无水甲醇配制碘-二氧化硫的溶液,并添加无水的吡啶或适宜的有机叔胺,以定量吸收反应产物HI,SO3。保障了滴定反应的准确定量进行。费休氏试剂中,仅碘与水作用的摩尔比1:1,作为化学计量反应定量的基础。反应完毕后,多余的游离碘呈现红棕色,即可确定为终点。
费休氏试液的配制
取碘,加入无水吡啶,,加入无水甲醇,冰浴冷却,通入干燥二氧化硫,加无水甲醇,暗处放置24小时。也可以使用稳定的市售费休氏试液。
费休氏试液的标定-----费休氏容量滴定测定法
库伦滴定测定法
注意事项
仪器要求
所用仪器应该<b>干燥,避免空气</b>中的水分侵入,测定操作可以在干燥处进行
适用范围
易与<font color="#f44336">I2和S02</font>反应的药物<font color="#d32f2f">不适用</font>
费休氏试液的选用:不同的<font color="#d32f2f">全自动水分测定仪</font>,对费休氏试液的要求也可能不同。所以,使用时,要根据试剂说明,正确使用
费休氏试液的安全处置:费休氏试液<font color="#f44336">有毒性</font>,<font color="#f44336">稳定性差</font>,<font color="#e57373">保存期短</font>,含吡啶的费休氏试液有恶臭。要特别注意。
炽灼残渣检查法
适用对象
<font color="#f44336">有机药物或挥发性无机药物</font>
原理
炽灼残渣:有机药物,挥发性无机药物,在硫酸条件下进行炭化和炽灼后,残留的硫酸盐灰分
方法
取供试品,置已经炽灼恒重的坩埚中,称定,加硫酸,缓缓低温加热直至完全<b>炭化</b>,放冷;加硫酸,低温低热至硫酸蒸气除尽后,炽灼完全<b>灰化</b>,移至干燥器,放冷,称定,炽灼至恒重。
注意事项
供试品的取用量:应根据炽<b>灼残渣限量和称量误差决定</b>。样品量过多,<b>炭化和灰化</b>时自太长;样品量过少,称量误差増大。一般应使炽灼残渣量为1~2mg,残渣限量一般为0.1%~0.2%当限量为0.1%,取样量约1g;限量为0.05%,取样约2g;限量为1%以上者,取样可在 1g 以下。
炭化过程控制:ChP 仍然规定为<b>先炭化后再加硫酸炽灼</b>,易导致灰化困难,时间延长,不易恒重。为了避免供试品在炭化时,骤然膨胀而逸出,可采用将<b>坩埚斜置</b>方式,<b>缓缓加热</b>,直至完全灰化(不产生烟雾)。<br>在进行高温炉内炽灼操作前,务必<b>蒸发除尽硫酸</b>,以免硫酸蒸气腐蚀炉膛,造成漏电事故。除尽硫酸蒸气,应<b>低温加热</b>,以防由于温度过高,供试品飞溅,而影响测定的结果。<br>
坩埚的选用:通常使用<b>瓷坩埚</b>。含<font color="#f44336">氟</font>的药品对瓷坩埚有<font color="#f44336">腐蚀</font>,应采用<b>铂坩埚</b>。瓷坩埚编号,可采用蓝墨水与 FeCl3 ,溶液的混合液涂写、烘烤、恒重后使用。
炽灼温度与恒重操作:不同国家药典所规定的炽灼温度也有所不同。ChP2015为700~800℃<br>一些<font color="#f44336">重金属(如铅)</font>于<font color="#d32f2f">高温</font>下易<font color="#f44336">挥发</font>,如需将炽灼残渣留作重金属检查,则<font color="#f44336">炽灼温度</font>必须控制在500~600℃。
ChP2015与BP2015均要求:炽灼残渣达恒重后,进行限量计算。而USP38与JP16均规定:残渣在限度外时,才要求炽灼至恒重后,进行限量计算。<b>炽灼至恒重的第二次称重应在继续炽灼30分钟后进行。</b>
第四节 药物中特殊杂质的检查
现代色谱分离
有杂质对照品的鉴定法
无杂质对照品的联用鉴定法
基因毒性杂质的检查
第四章 药物的含量测定与分析方法的验证
定量分析法的分类
容量分析法
直接滴定
间接滴定
生成物滴定法
剩余量滴定法
光谱分析法
紫外-可见分光光度法
对照品比较法
吸收系数法
计算分光光度法
比色法
荧光分光光度法
色谱分析法
高效液相色谱法
内标法
外标法
气相色谱法
标准溶液加入法
第五章 体内药物分析
体内样品的处理
体内样品分析方法与方法验证
概述
体内药物分析,也称生物分析,是指体内样品中药物及其代谢产物或内源性生物活性物质的定量测定
体内样品性质特点
采样量少,且不易重新获得。ml-ul
待测物浓度低,ug/ml
干扰物质多,血样和组织中的蛋白质
体内药物分析的特点
样品需要经纯化浓集,或者化学衍生化处理
对分析方法的灵敏度和专属性要求较高
分析工作量大,数据处理和结果阐明繁杂
常用药物体内样品的制备与储存
体内样品的种类包括体液,组织,排泄物等,最常用的是血浆或血清--因为他们可以较好的体现药物浓度和治疗作用之间的关系
体内样品的采集与制备
血样
血药浓度常指血浆或血清中药物的浓度,而不是全血药物浓度。
血浆和血清可以任意选用,血浆中药物的浓度可以反应药物在体内作用部位的状况
采集--静脉,1-5ml
制备
血浆50-60%--抗凝剂,离心,淡黄色上清液
血清20-40% 静置,离心,上层淡黄色液体
全血,抗凝剂,不离心
尿样
药物可以以原型或者代谢物的形式从尿中排出,用于药物的物质平衡,排泄途径,尿清除率研究
特点---原形,代谢物及缀合物。药物浓度高,收集量大
采集--自然排尿--规定时间段
贮藏--加入适当防腐剂
尿液浓度变化较大,应测定一定时间内排入尿液中的药物总量,测定在规定时间内采集的尿液体积和浓度
唾液
当药物的唾液浓度与血浆游离浓度密切相关时,在TDM时可以用S代替P
采集--漱口后15min,安静状态,自然流出的唾液(腮腺,舌下腺,颌下腺)
制备--离心,上清液
组织
制备
制成水性基质匀浆溶液
处理
蛋白沉淀法--蛋白沉淀剂--三氯乙酸
酸水解或碱水解法--
酶水解法---蛋白水解酶
储存和处理
冷藏与冷冻
血样:分离--硬质玻璃,聚乙烯管--冷冻长期保存,4°短期保存
尿样:冷藏,加防腐剂,冷冻
唾液:4°以下,冷冻保存时,解冻后充分搅匀后再用
去活性
采样后立即终止酶的活性,常用方法--液氮中快速冷冻,微波照射,匀浆,沉淀,加入酶活性阻断剂,抗氧化剂,调节pH,样品煮沸等
体内样品的处理
前处理--分离,浓集,改性---为药物的测定提供条件
前处理是体内药物分析过程中重要且繁复的工作
体内样品处理的目的
去使待测组分游离
药物进入体内会与血浆蛋白结合,或转化为代谢产物。---使待测药物释/代谢物释放,测定药物/代谢物总浓度
满足测定方法的要求
样品介质组成复杂:分离
待测组分浓度低--浓集
改善分析环境
防止仪器污染,劣化(去蛋白)
提高测定灵敏度/选择性--化学改性(衍生化)
常用的体内样品处理方法
去除蛋白质
溶剂沉淀法
加入与水相混溶的有机溶剂,蛋白质分子间引力增加而聚集,水化膜脱水,蛋白质沉淀,药物释放
中性盐析出法
中和蛋白质电性,改变分子间电排斥作用而凝聚,同时使蛋白质水化膜脱水而析出沉降
强酸沉淀法/加入强酸
与蛋白质阳离子形成不溶性盐沉淀---当溶液pH低于蛋白质等电点,蛋白质以阳离子形式存在,可与酸根阴离子成盐
加入重金属盐
与蛋白质阴离子形成不溶性盐沉淀
分离与浓集
液相萃取法LLE
液-液提取法
是利用待测药物与内源性干扰物质的分配系数不同而进行的液相分离技术。利用亲水和亲脂性质的不同。
溶剂选择
对药物的分子形式可溶,电离形式不容
沸点低,易挥发
与水不混溶
无毒,不易燃烧
较高的化学稳定性和惰性
不影响紫外检测
溶剂用量
适当,1:1;1:5
水相pH
根据药物的pKa调节。碱性药物的最佳pH高于pKa1-2个pH单位,酸性药物最佳pH低于pKa1-2个单位
提取操作
一般只提取一次
若提取回收率较低,提取2-3次
脂溶性干扰物,可进行小体积水溶液反提取
固相萃取法 SPE
原理
将不同填料作为固定相,当含有药物的体内样品溶液经过时,由于受到吸附,分配,离子交换或其他亲和力作用,产生药物被保留或洗脱的结果,从而达到分离和浓集的作用
自动化固相萃取法
优点
重现性好,批量处理,富集互溶净化,避免乳化
缺点
小柱成本高,方法开发
固相微萃取技术 SPME
选择不同的石英纤维涂层,使目标化合物能吸附在涂层上,而干扰化合物不吸附。非极性药物选择非极性涂层
优点---操作时间短,样品量小,无需提取溶剂,重现性好,适于分析挥发性与非挥发性物质。
微透析MD
在不破坏或很少破坏生物体环境的前提下,对生物体细胞液的内源性或外源性物质进行连续取样和分析的新技术
时间分辨性
空间分辨性
超滤法
是一种膜分离技术,按照截留分子量选择半透膜,可分离10-1000kD的可溶性生物大分子,无化学试剂,无相变化
简便,游离药物分析首选方法,尤其适合TDM
缀合物水解
测定尿药总量,水解缀合物,趋向于直接测定缀合物
药物或其Ⅰ相代谢产物与体内的内源性物质结合的产物称为Ⅱ相代谢产物或者缀合物
酸水解法
简便,快速,水解过程中发生药物分解,专一性差
酶水解法
葡萄糖醛酸苷酶或/和硫酸酯酶
专属性强,时间较长可引入粘蛋白
溶剂解法
溶剂萃取过程中缀合物直接分解(尤其是硫酸酯)
化学衍生化
气相色谱GC
提高药物挥发性,增加药物的稳定性,生成非对映异构体
高效液相色谱HPLC
提高检测灵敏度,改善色谱分离
体内样品分析方法与方法验证
分析方法的选择
色谱分析法
免疫分析法
生物学方法
分析方法建立的一般程序
色谱条件的筛选
色谱条件的优化
实际样品的测试
分析方法的验证
生物分析必须汾河药物非临床试验质量管理规范GLP或者药物临床试验质量管理规范GCP的相关要求
完全验证---部分验证---交叉验证
名词解释
对照标准物质
生物样品中药物及其代谢产物的测定需使用含对照标准物质的校正样品和质量控制样品作为参比,一般对照标准物质的来源有
药典标准物质
从行业内具有相关资源,公认信誉度良好的公司购买的对照标准物质
由分析实验室或者其他非商业盈利机构合成和标化的有明确纯度的标准物质
基质效应
是指在样品测试过程中,由于待测物以外的其他物质的存在,直接或间接影响待测物响应的现象,在质谱检测中,基质效应会改变待测物质的离子化效率,引起测定物检测信号抑制或增强,除正常基质外,还应关注其他样品的基质效应。
评价方法
柱后灌注法
提取后加入法
质控样品QC样品
在空白基质中加入已知量待测物质对照标准物质制成的样品,用于监测生物分析方法的效能和评价每一分析批中试验样品分析结果的完整性和正确性。
分析批
包括试验样品,适当数量的校正标样和QC样品的一个完整系列。一个分析批连续测量不宜超过3天
分析方法的验证
选择性
标准曲线与定量范围
定量下限
精密度与准确度
样品稳定性
提取回收率
分析过程的质量控制
未知样品浓度超限的处理
药物内源性物质的测定
微生物/免疫学方法的验证
药剂学
第十一章 固体制剂的单元操作
固体制剂是以固体状态存在的剂型总称。占有率≥70%
粉碎
粉碎是指借助机械力或其他方法,将大块固体物料破碎和展磨成块状,细粉或者超细粉的过程
粉碎的意义
增加表面积,有利于提高难溶性药物的溶出速度以及生物利用度
有利于各层分混合均匀
改善制剂的质量,有利于提高固体药物在液体,半固体,气体中的分散度
有助于天然药材中提取有效成分的溶解度,提高有效成分的提取效率
原理
借助外加作用力破坏分子间的内聚力
粉碎后,表面积增大,因此粉碎实际上是机械能转变为表面能的过程
粉碎方法
开路与循环
开路粉碎
物料进入粉碎机粉碎后直接得到产品
操作简单,粒度分布宽,适用于粗碎或粒度要求不高
循环粉碎
物料进入粉碎机粉碎后经过分级,在返回粉碎机粉碎,得到产品,循环粉碎
动力消耗相对低,粒度分布窄,适用于粒度要求较高的粉碎
闭塞与自由
闭塞粉碎
不能及时分离物料,影响继续粉碎
软垫,效率低,常用于间歇操作
自由粉碎
可以及时分离物料,不影响继续粉碎
效率高,常用于连续操作
区别是达到粉碎要求的粉末不能/能及时排出,从而影响。不影响继续粉碎的操作
单独与混合
单独粉碎
贵重物料,刺激性药物,氧化性和还原性等必须单独粉碎
混合粉碎
两种以上的物料共同粉碎的操作
避免粘性药物单独粉碎的困难
混合和粉碎结合进行--性质,硬度相近的
串研法,串油法
干法与湿法
干法粉碎
将物料经过适当的干燥处理后,水分含量下降至一定程度进行粉碎的方法,一般低于5%
湿法粉碎
将适量的液体添加至药物中进行研磨粉碎
避免粉尘飞扬,降低药物的黏附性
水飞法--加液研磨法
水飞法
是将药物和水共同置于研钵或球磨机中一起研磨,使细粉漂浮于液面或者混悬于水中,然而将此混悬液倾出,余下的粗料加水反复操作,至全部药物研磨完毕,所得混悬液合并,沉降倾去上层清液,将湿法干燥,粉碎得极细粉。
低温粉碎
利用药物在低温下脆性增加,韧性和延伸性降低得性质,以提高粉碎效率方法
适用范围
热敏感,软化温度低,易于成饼得药物,
优缺点
可获得更细得粉末,且可保存物料中得香气及挥发性有效成分,提高药效
较普通粉碎得费用高,设备较为困难
粉碎设备
研钵
球磨机
冲击式粉碎机
万能粉碎机
震动磨
流能磨
高压均质机等
制备微乳,脂质体,混悬剂等
原则
根据目的和剂型控制粉碎程度
粉碎过程中及时过筛,以免过度粉碎
粉碎毒性药物或刺激性较强得药物时,应注意劳动保护,以免中毒。粉碎易燃易爆药物时,要注意防火防爆
中药材得药用部分必须全部粉碎应用
分级/筛分
筛分法
利用网性工具使粗粉和细粉分离得操作
目的是为了获得较均匀得离子群
药筛得种类和规格
冲眼筛
编织筛
第十二章 固体制剂
常用辅料
稀释剂/填充剂
淀粉
蔗糖
糊精
乳糖
预胶化淀粉
MCC
无机盐
糖醇类
润湿剂
水
乙醇
粘合剂
淀粉浆
纤维素衍生物
甲基纤维素
羟丙纤维素
羟丙甲纤维素
羧甲基纤维素
PVP
明胶
乙基纤维素
崩解剂
干淀粉
羧甲淀粉钠
低取代羟丙纤维素
交联羧甲纤维素
交联聚维酮
泡腾剂得崩解剂
表面活性剂
润滑剂
硬脂酸镁
微粉硅胶
滑石粉
氢化植物油
聚乙二醇
月桂醇硫酸钠/镁
色香味调节剂
着色剂
矫味剂
第五节--胶囊剂
子主题
粘膜给药
肺黏膜给药
气雾剂的处方设计
溶液型气雾剂
如果药物能够溶解于抛射剂中克制成溶液型气雾剂
辅助性溶剂既可以和抛射剂混溶,又可以增加药物的溶解度,这类溶剂被称为潜溶剂--潜溶剂的作用是要让药物溶解得到澄明均相溶液
潜溶剂--乙醇,聚乙二醇,丙二醇,甘油,乙酸乙酯,丙酮等
但要注意刺激性和毒性
乙醇,甘油可以用于口腔吸入或鼻腔气雾剂
抛射剂的用量越大,喷出的液滴越细
注意的问题
抛射剂与潜溶剂对药物溶解度与稳定性的影响
喷出液滴的大小与表面张力对用药部位的影响
抗氧剂,防腐剂,潜溶剂对用药部位的刺激性
吸入剂中各种附加剂是否能在肺部代谢或滞留
混悬型气雾剂
先将药物进行微粉化处理,然后制成混悬液,再进行分装,添加适量助悬剂
制备有一定的难度,颗粒变大,聚集,结块,堵塞阀门
为防止颗粒变大,选择再抛射剂中溶解度较小的衍生物
乳剂型气雾剂
先将药物溶剂在水性或者油性介质中,然后再制成均匀的乳剂,再进行分装,乳化剂的选择很重要
气雾剂的制备工艺
填充抛射剂
压灌法
冷灌法
乳剂型含水分的气雾剂不适用于此法进行罐装
气雾剂的质量评定
递送剂量均一性
喷出总量
每瓶总揿次检查
每揿喷量
微细粒子剂量
粒度
微生物限度
装量
每揿主药含量
无菌检查
喷射速率
气雾剂的装置
气雾剂主要有药物,喷射剂组成,使用可通过定量阀门控制剂量,具有速效和定位作用,又被称为定量吸入气雾剂
MDI需要特定的装置,加压定量吸入器,pMIDs,由耐压容器,定量阀和驱动装置三部分组成,含药溶液,乳状液或混悬液与适宜的抛射剂共同封装于耐压容器中,患者按压驱动装置,药物与抛射剂以小液滴形式被释放出了,抛射剂迅速挥发,药物粒子被吸入肺中
MDI局限性
抛射剂氟利昂破坏大气臭氧层
需要耐压容器和阀门系统等机械设备
喷雾剂
喷雾剂不含抛射剂
特点
局部应用为主,喷射的液滴较粗,但可以满足临床需求
由于不是加压包装,制备方便,成本低
既有物化给药的特点,又避免使用抛射剂
装置
雾化器
使用氧气,加压气体,超声振动或者其他方法将药物溶液,乳液或混悬液分散为小液滴喷出,患者可以通过该装置的入口端直接吸入给药
手动泵
容器
质量评价
每瓶总喷数
每喷喷量
每喷主要含量
递送剂量均一性
微细粒子剂量
装量差异
装量
无菌
微生物限度
粉雾剂
分吸入粉雾剂,非吸入粉雾剂和外用粉雾剂
吸入粉雾剂
可以发挥局部及全身作用,具有靶向,高效,速效,毒副作用小等特点
大小小于10,大多数小于5
可加入附加剂改善粉雾剂的流动性
粉雾剂易吸潮,应注意储存条件
处方分类
仅含微粉化药物的粉雾剂、
、药物加适量附加剂改善粉末的流动性
一定比例的药物和载体混合均匀体
药物与载体及各种附加剂的混合体
为防止药物的聚集,增加其流动性,尝使用载体
常用载体--乳糖FDA唯一批准
药物与载体的内聚力适宜
考虑因素
药物
粒径的大小和形态球形粒子最佳
药物的流动性与分散性-
改善流动性---加入少量苯甲酸钠,硬脂酸镁,胶体硅等润滑剂和表面活性剂如波洛沙姆等抗静电剂
电性
粒子之间摩擦产生静电---表面活性剂可以消除静电,毒性小--泊洛沙姆
附加剂
主要作用提高粉末的流动性
表面活性剂,分散剂,润滑剂,抗静电剂
载体
在粉雾剂中起到稀释剂和改善微粉化后药物的流动性的作用
乳糖--常用载体,FDA唯一批准
此外蛋白稳定剂,肺部内源性物质也可以作为载体
给药装置
胶囊型给药装置
泡囊型给药装置---碟式吸入器
贮库型给药装置
供雾化器用的液体制剂
通过连续或者定量雾化器产生供吸入用气溶胶的溶液没混悬液,乳液
检查
递送速率和递送剂量
递送速率和递送剂量,微细粒子剂量,无菌检查
超声雾化器
压缩式雾化器
直肠粘膜给药
生理结构
乙状结肠到肛门20cm长的笔直部分
直肠粘膜结构与小肠基本相同,但是皱襞少,吸收面积小
直肠中ph接近中性或偏碱性,缓冲能力弱
吸收途径
经肝门系统
直肠上静脉---6cm
非经肝门系统
直肠中静脉,直肠下静脉,肛管静脉---2cm
影响直肠吸收的因素
影响吸收的生理因素
直肠内容物--栓剂的使用深度--ph的影响--病理状态
药物的理化性质
溶解度---大更容易吸收
粒度---以为溶解状态存在于栓剂中的药物,其粒度大小影响释放,溶解及吸收,粒径越小,越容易溶解,吸收越快
解离度--未解离的药物容易透过肠道粘膜吸收入血,未解离的分子越多,吸收越快
弱酸性,弱碱性药物容易吸收,可以使用缓冲剂改变直肠部位ph,增加非解离性药物的比例
基质和附加剂的理化性质
基质的种类
发挥全身作用,选择与药物溶解性相反的基质,有利于药物的释放,增加吸收。
发挥局部作用,通常药物不需要吸收,基质应缓慢融化以延缓药物释放--局部作用通常半小时开始起效,至少持续4h
附加剂
表面活性剂
栓剂
局部作用,全身作用
性状---鱼雷形,鸭嘴形
栓剂的基质
理想栓剂的基质
与药物混合不发生反应,不妨碍主要的作用和含量测定
对黏膜吸收无刺激,无毒,和致敏性
室温下具有适宜的硬度和韧性,体温下易与软化,融化或溶解
释药速度符合治疗要求
酸值,碘值,皂化值满足要求
具有润湿或乳化能力,混入较多的水
具有润湿或乳化能力,混入较多的水
适用于挤压成型法及模制成型法制备
常用基质
油脂性基质
可可豆脂
半合成或全合成脂肪酸甘油酯
氢化植物油
水溶性基质
甘油明胶
凡与蛋白质能产生配伍变化的药物,如鞣酸,重金属盐等,均不能用甘油明胶作基质
聚乙二醇
PEG
泊洛沙姆
聚氧乙烯单硬脂酸酯
常用附加剂
吸收促进剂
吸收阻滞剂
硬化剂
增稠剂
栓剂的制备
挤压成型法
冷压法
模制成型法
热熔法
栓剂的置换价
第六章
芳酸类非甾体抗炎药物的分析
主要理化性质
酸性
较强酸性--水杨酸,阿司匹林,双水杨酯,二氟尼柳
较弱酸性--双氯芬酸,布洛芬,酮洛芬,萘普生,吲哚美辛
酸性很弱--吡罗昔康,美洛昔康,尼美舒利,对乙酰氨基酚
原料药可在中性乙醇或甲醇,丙酮等水溶性有机溶剂中,用氢氧化钠直接滴定
水解性
酯键
阿司匹林,双水杨酯
酰胺键
吲哚美辛,吡罗昔康,美洛昔康,尼美舒利,对乙酰氨基酚
水解反应及其产物的特性反应,用于鉴别----水解反应可快速,定量进行,用剩余碱量法测定含量
吸收光谱特性
苯环--特征取代基
紫外特征光谱--鉴别,含量均匀度,溶出度,释放度
红外特征光谱--鉴别
基团/元素特性
酚羟基与三价铁离子配位显色---鉴别
二苯甲酮与苯肼缩合显色---鉴别
酮洛芬
硫与醋酸铅生成黑色硫化铅---鉴别
美洛昔康
鉴别实验
与三氯化铁反应
凡事具有酚羟基的药物都具有此反应
水杨酸反应
中性或弱酸性条件下进行
灵敏度高,在稀溶液中进行
水杨酸类
阿司匹林--煮沸水解---水杨酸--紫瑾色
双水杨酯---氢氧化钠--水杨酸--紫色
二氟尼柳--乙醇--(水中溶解性差)--深紫色
酚羟基反应
对乙酰氨基酚
蓝紫色
烯醇式羟基
吡罗昔康
三氯甲烷---玫瑰红色
美洛昔康
三氯甲烷--淡紫红色
缩合反应
酮洛芬
酮洛芬的二苯甲酮,在酸性条件下与二硝基苯肼生成橙色缩合产物沉淀
重氮化-耦合反应
芳香第一胺类鉴别
凡事分子中含有芳伯氨基或潜在芳伯氨基的药物均可反应
对乙酰氨基酚
对乙酰氨基酚--酸性条件--对氨基酚--游离芳伯氨基在亚硝酸钠试液中重氮化---重氮盐与碱性-β萘酚偶合生成红色偶氮化合物
水解反应
阿司匹林+碳酸钠----水解---水杨酸钠和醋酸钠---酸化--白色水杨酸沉淀---发醋酸的臭气
双水杨酯---氢氧化钠--水杨酸盐的鉴别反应
FeCl3--紫色
稀盐酸,析出白色沉淀,沉淀在醋酸铵上溶解
元素反应
氯的鉴别---与碱共热产生氯化物,显氯化物的鉴别反应
双氯芬酸钠
碳酸钠炽灼炭化
硫的鉴别--经高温分解产生硫化氢气体,与醋酸铅棉花生成硫化铅黑色沉淀
美洛昔康
光谱法
紫外-可见分光光度法
最大吸收波长
最大与最小吸收波长
吸光度法
吸光度比值法
红外分光光度法
化学结构复杂,差异小的药物的鉴别---专属性强
色谱法
薄层色谱法
高效液相色谱法
有关物质的检查
阿司匹林与双水杨酯
阿司匹林
游离水杨酸
游离水杨酸对人体有毒性,酚羟基会被空气,逐渐氧化生成淡黄,红棕至深棕色醌型化合物
采用1%冰醋酸甲醇溶液制备供试品溶液,临用新制,防止阿司匹林水解,
HPLC检查,
有关物质
不加校正因子的主成分自身对照法
HPLC
双水杨酯
游离水杨酸
水相萃取比色法
三氯甲烷溶解----硝酸铁的稀硝酸溶液--测吸光度
对乙酰氨基酚
有关杂质
对氨基酚,对氯苯乙酰胺等
对氨基酚及有关物质---HPLC---离子对色谱法
离子对试剂---四丁基氢氧化铵
对氯苯乙酰胺---HPLC---磷酸盐缓冲液-甲醇
含量测定
原料药测定法
直接滴定法
-COOH,可以直接用碱滴定
缺点--酸性杂质干扰,酯键水解干扰
适用范围--仅用于合格原料药测定,不适用于制剂含量的测定
应用---针对具有游离羧基的原料药,水杨酸,双水杨酯,二氟尼柳,等。选择合适的溶剂和指示剂
注意事项--规范操作,不断旋摇,快速滴定--减少水解产生的误差
水解后剩余量滴定法
适用于含有酯的结构药物
优点--消除了酯键水解的干扰
缺点--酸性杂质干扰
做空白实验
反滴定法
美洛昔康
和羧基共轭的烯醇式羟基,具有羧酸性质,甲醇,乙醇,丙酮,水中难溶,定量过量的氢氧化钠滴定液溶解后,用盐酸滴定液回滴定
药物制剂分析法
直接紫外分光光度法
色谱柱分配-紫外分光光度法
高效液相色谱法
第七章
苯乙胺类拟肾上腺素药物的分析
结构与性质
结构
肾上腺素--异丙肾上腺素---重酒石酸去甲肾上腺素--盐酸去氧肾上腺素--重酒石酸间羟胺--盐酸克伦特罗
性质
酚羟基特性
邻苯二酚,酚羟基
与金属离子配位显色
易被氧化
弱碱性
烃氨基侧链
游离碱难溶于水,易溶于有机溶剂,其盐可溶于水
旋光性
手性碳原子
吸收光谱特性
苯环特征吸收带,254nm为中心红移蓝移
鉴别实验
与铁盐反应
酚羟基--三氯化铁--绿色/--碱性条件下--紫红色--被氧化
用于肾上腺素,盐酸异丙肾上腺素,重酒石酸去甲,盐酸去氧,盐酸多巴胺,硫酸沙丁胺醇的鉴别
沙丁胺醇--紫色
盐酸去氧肾上腺素---反应不同
紫色
与甲醛-硫酸反应
酚羟基--甲醛硫酸--有色化合物--醌式结构--氧化
还原性试验
酚羟基--碘-过氧化氢--铁氰化钾--有色化合物
区别重酒石酸去甲肾上腺素
氨基醇的双缩尿反应
氨基醇--硫酸铜--碱性--紫色络合物
盐酸麻黄碱
盐酸去氧肾上腺素
脂肪伯胺的Rimini试验
脂肪伯胺--亚硝基铁氰化钠-丙酮-碳酸氢钠--60℃,水浴--紫红色
鉴别脂肪伯胺的专属反应
所含丙酮必须不含有甲醛成分
重酒石酸间羟胺
吸收光谱特性
紫外
红外
特殊杂质与检查
酮体杂质的检查
光学纯度的检查
有关物质的检查
含量测定
非水溶液滴定法
非水溶液滴定法
非水溶液滴定法的种类
非水酸量法
非水碱量法
有机弱碱
有机碱的无机盐--氢卤酸盐,磷酸盐,硫酸盐
有机碱的有机盐
非水溶剂的种类
酸性溶剂
冰醋酸
碱性溶剂
DMF-二甲基酰胺
两性溶剂
甲醇
惰性溶剂
氯仿,甲苯,丙酮
适用范围与溶媒的选择
适用范围
PKb>8有机弱碱或盐的药物
溶媒的选择--提高样品碱性;溶解样品及产物溶剂;与样品及滴定剂三者之间无副反应
8-10------冰醋酸
10-12 ----冰醋酸+醋酐,硝基甲烷+醋酐
12---醋酐
加入醋酐时,主义防止氨基苯乙酰化,所以在冰醋酸溶解样品后,应放冷后再加醋酐
基本原理
强酸制弱酸,反应式见书
酸根的影响
各种酸在水溶液中因为拉平效应酸性差不多;而在冰醋酸中高氯酸酸性最强,所以用高氯酸滴定
测定有机碱的卤酸根盐时
加入卤化汞冰醋酸---使其生成在醋酸中难解离的卤化汞,以消除氢卤酸对滴定的干扰和不良影响
测定有机碱的硝酸盐---释放的硝酸破坏指示剂--电位法指示终点或加入Vc
测定有机碱的硫酸盐
硫酸沙丁胺醇的滴定---硫酸在冰醋酸中是一元酸,化学剂量关系1:1,酸性强,溶剂中加入大量醋酐,并用电位法指示终点
滴定剂的稳定性
冰醋酸具有挥发性,膨胀系数大,若滴定样品与标定滴定液的温度差未超过10℃,将高氯酸滴定液的浓度加以校正。超过10℃重新标定。
盐酸异丙肾上腺素原料药的测定,盐酸布比卡因的含量测定
溴量法
盐酸去氧肾上腺素,重酒石酸间羟胺原料药
原理
药物分子中的苯酚,其邻位和对位活泼的氢可以和溴发生溴代反应,然后用硫代硫酸钠滴定剩余的溴,测得含量
溴代反应
注意事项
置于碘瓶---防止溴和碘的挥发
用空白试验校正,空白试验可以减少溴和碘逸失带来的误差
溴滴定液--是溴酸钾加溴化钾,然后在酸性条件下反应生成溴
溴标准滴定液不能加入太多--因为有氧化作用,会把苯酚氧化,及破坏药物分子
在酸性--HCL--中进行
亚硝酸钠滴定法
具有芳伯氨基,及潜在芳伯氨基的药物
盐酸克伦特罗---重氮化反应--重氮盐--永停滴定法
具体见第八章含量测定
紫外分光光度法
盐酸甲氧明注射液
杂质/辅料对吸收无干扰
百分吸收系数法
注意单位
比色法
盐酸克林特罗栓
重氮化偶合显色或与亚铁离子配文显色
氨基磺酸铵分解剩余亚硝酸--偶合剂与亚硝酸反应也能显色干扰测定
动物组织中盐酸克伦特罗残留的测定
液液萃取
乙酸乙酯
提取溶剂
碳酸钠溶液
提供碱性环境---盐酸克伦特罗显弱碱性--在碱性环境中游离多
盐酸溶液
成为盐酸盐,溶于水中
涡旋--混匀
离心--分离分层
固相萃取净化
阳离子交换小柱
酸性环境下,克伦特罗呈阳离子与小柱结合,酸性或中性物质可以用水和甲醇淋洗除去
依次用水和甲醇淋洗柱子
除去中性或酸性物质
氨化甲醇洗脱
实现待测成分的净化
衍生化--硅烷化
第八章
对氨基苯甲酸酯和酰苯胺类局麻药物的分析
对氨基苯甲酸酯类局麻药物
普鲁卡因
苯佐卡因--盐酸普鲁卡因--盐酸丁卡因--盐酸氯普鲁卡因---盐酸普鲁卡因胺
先苯胺类局麻药物
利多卡因
盐酸利多卡因--盐酸布比卡因-盐酸罗哌卡因
主要理化性质
水解性
对氨基苯甲酸酯中含有酯键--易水解
特殊杂质--PABA,BABA,2,6-二甲基苯胺,4-氨基-2氯苯甲酸
酰苯胺利多,布比,罗哌卡因由于空间位阻--较难水解
芳伯氨基特性
对氨基苯甲酸酯类药物的结构中含有芳伯氨基---重氮化-偶合反应
酰苯胺类由于空间位阻影响,较难水解,重氮化-偶合反应不适用
弱碱性
脂烃胺侧链为叔胺氮原子,具有一定碱性
和生物碱称点击发生反应,生成沉淀可鉴别
在水溶液中不能用标准酸直接滴定,只能在非水溶液中滴定
与重金属离子反应特性
利多,布比,罗哌卡因酰胺基上的氮可以铜离子,钴离子生成有色配位化合物沉淀
光谱吸收特性
苯环,取代基,脂烃胺侧链
紫外吸收光谱,红外光谱
其他特性
游离碱多为碱性油状液体或低熔点固体,游离碱难溶于水,易溶于有机溶剂;可以成盐,有机碱盐易溶于水,难溶于有机溶剂
鉴别实验
重氮化-偶合反应--芳香第一胺类鉴别反应
苯佐卡因,盐酸普鲁卡因,盐酸氯普鲁卡因,盐酸普鲁卡因胺都能发生此反应
盐酸丁卡因不具备芳伯氨基,但分子结构中的芳香仲胺在酸性溶液中与亚硝酸钠反应,生成白色沉淀,鉴别
与金属离子反应
盐酸利多卡因的鉴别
与铜盐反应
水,碳酸钠,硫酸铜--蓝紫色--三氯乙烷--黄色
与钴盐反应
氯化钴--亮绿色
在酸性溶液中,与氯化钴反应,生成亮绿色细小钴盐沉淀
与汞盐反应
利多卡因--硝酸--硝酸汞--黄色
对氨基苯甲酸酯--硝酸--硝酸汞煮沸--红色/橙黄色
盐酸普鲁卡因胺的鉴别
羟肟酸铁盐反应
样品==煮沸--三氯化铁--煮沸---紫红色---暗棕色--棕黑色
水解产物
盐酸普鲁卡因的鉴别
专属反应
水,氢氧化钠,白色沉淀,加热--油状物--加热--蒸气(红色石蕊试纸变蓝)--盐酸--白色沉淀--过量溶解
苯佐卡因的鉴别
氢氧化钠---乙醇--碘--黄色沉淀--碘仿臭气
制备衍生物测熔点
盐酸丁卡因的鉴别
沉淀--过滤--洗涤--干燥--测熔点
吸收光谱特征
紫外吸收光谱
规定浓度的供试液在最大吸收波长,测定吸光度
盐酸布比卡因
盐酸普鲁卡因胺片
红外吸收光谱法
盐酸普鲁卡因的鉴别
特殊杂质与检查
对氨基苯甲酸类杂质的检查
杂质来源--酯键水解
盐酸丁卡因--对丁基苯甲酸
盐酸普鲁卡因--对氨基苯甲酸
盐酸氯普鲁卡因--4-氨基-2-氯苯甲酸
盐酸普鲁卡因中杂质对氨基苯甲酸的检查
HPLC杂质对照品法
庚烷磺酸钠---中性粒子对,与非极性固定相作用强,改善分离效果
盐酸氯普鲁卡因主射液有关物质检查
LC-MS
液相色谱质谱联用
光解产物--盐酸羟基普鲁卡因
酰苯胺类局麻药中2,6-二甲基苯胺及其他杂质的检查
酰胺键水解
盐酸利多卡因及其注射液
HPLC
主成分自身对照法
杂质对照品法
盐酸罗哌卡因及其注射液,注射用盐酸罗哌卡因
盐酸罗哌卡因的光学纯度检查
手性碳原子--S为有效体,R有心脏毒性
HPLC\
毛细管电泳法--USP34-NF29
含量测定
亚硝酸钠滴定法
具有芳伯氨基或潜在芳伯氨基的药物
可以与亚硝酸钠反应,生成重氮盐
测定的主要条件
反应速度与药物结构的关系
苯环上有吸电子基团,反应速度加快
苯环上有供电子基团,反应速度减低
加入适量KBr加速反应
溴化钾与盐酸反应生成HBr,生成的亚硝酰溴比亚硝酰氯反应更快,加快重氮化反应速度
酸的种类及其浓度
不同酸中重氮化反应的速度不同,氢溴酸最大
加入过量的盐酸和溴化钾
酸度加大,重氮化反应速度加快
重氮盐在酸性溶液中稳定
酸度加大,反应向左进行,防止生成偶氮氨基化合物影响测定结果
酸度过大,又可阻碍芳伯氨基的游离,反而影响重氮化反应速度
在太浓的盐酸中还可使亚硝酸分解
反应温度
室温条件下滴定
温度高,反应速度快,每升高10℃加快2.5倍,温度太高,可使重氮盐分解
测定一般在低温下进行,由于低温时反应太慢,经试验,可在室温下进行,其中15℃以下结果较准确
滴定速度
先快后慢
尖端插入液面2/3下,一次大部分放下,缓慢滴定,最后一次加入,搅拌1-5分钟
指示终点的方法
永停滴定法
电极反应--氧化还原反应
缺点--电极容易钝化
电位法
外指示剂法
KI-淀粉糊剂或试纸
缺点--未到终点的时候可能误判
多次外试导致样品损失,误差
内指示剂法
中性红不可逆指示剂
盐酸普鲁卡因,盐酸克伦特罗
非水溶液滴定法
盐酸布比卡因的含量测定
弱碱性
见第七章含量测定
紫外分光光度法
HPLC法
第十章 巴比妥类及苯并二氮杂卓类镇静催眠药物的分析
巴比妥类
巴比妥类药物的主要理化性质
巴比妥--苯巴比妥--司可巴比妥--硫喷妥钠
弱酸性
巴比妥类药物分子结构中都有1,3-二酰亚胺基团,能发生酮时和烯醇式的互变异构,在水溶液中可以发生二级电离
本类药物具有弱酸性,可与强碱反应生成水溶性的盐类,一般为钠盐---可以用于巴比妥类药物的分离
水解反应
巴比妥类药物分子 结构中含有酰亚胺结构,与碱液共沸即水解,释放出氨气。可以使湿润的红色石蕊试纸变蓝
本类药物的钠盐,在吸湿的情况下会发生水解,注射剂制成粉针剂,现配现用
与金属离子的反应
丙二酰脲或酰亚胺基团在合适pH的溶液中可与某些重金属离子,反应呈色或产生有色沉淀
与银盐的反应
碳酸钠--碱性环境---生成钠盐溶解--硝酸银---生成可溶性一银盐--加入过量硝酸银---生成二银盐白色沉淀
与铜盐的反应
吡啶溶液--碱性环境---烯醇式异构体与铜吡啶试液反应---有色配位化合物
巴比妥类药物呈紫堇色------含硫巴比妥呈现绿色
与钴盐的反应
在碱性溶液中可与钴盐反应,生成紫堇色配位化合物
反应在无水条件下比较灵敏,生成的有色产物也比较稳定。
异丙胺---无水碱性环境
与汞盐的反应
与硝酸汞或氯化汞溶液反应,可生成白色汞盐沉淀,在氨试液中分解
酸性条件下进行
与香草醛的反应
vanilin
丙二酰脲基团中的氨比较活泼,可与香草醛在浓硫酸存在下发生缩合反应,生成红棕色产物
巴比妥---香草醛--硫酸--水浴加热--红棕色----放冷--乙醇--紫色--蓝色
紫外吸收光谱特性
巴比妥类药物的紫外吸收光谱随着其电离级数不同发生变化
0.05mol/l硫酸溶液--未电离--无吸收峰
pH9.9缓冲溶液--一级电离--240nm
0.1mol/l氢氧化钠--pH13--二级电离--25nm吸收峰红移--向波长更长的方向移动
硫代巴比妥类药物的紫外吸光谱则不同,在酸性或碱性溶液中均有比较明显的紫外吸收
薄层色谱行为特征
GF254薄层板---
H--不含黏合剂
G--含黏合即
F--荧光
观察荧光淬灭
显微结晶
巴比妥药物本身或者反应产物--特殊晶型--适用于生物群体样本中微量药物的检验
鉴别反应
丙二酰脲反应
巴比妥类药物母核的反应
银盐反应
铜盐反应
特征基团反应
利用硫元素的鉴别实验
硫代巴比妥类分子中含有硫元素--氢氧化钠--铅离子--生成白色沉淀--加热--沉淀转变为硫化铅--黑色沉淀-
可以供硫代巴比妥与巴比妥类鉴别
利用不饱和取代基的鉴别试验
丙烯基--不饱和基团--发生加成反应--氧化反应
与碘试液的反应
棕黄色在5分钟内消失
与溴试液反应--溴褪色
与高锰酸钾的反应
将紫色高锰酸钾还原为棕色的二氧化锰
利用芳环取代基的鉴别试验
硝基化反应
含有芳香取代基的巴比妥类药物,与硝酸钾和硫酸共热,可发生硝基化反应--生成黄色硝基化合物
苯巴比妥
与硫酸-亚硝酸钠的反应
苯巴比妥--硫酸-亚硝酸钠反应--生成橙黄色产物---随机变为橙红色
区别苯巴比妥和其他不含芳环取代基的巴比妥类药物
与硫酸-甲醛的反应
苯巴比妥与甲醛-硫酸反应,生成玫瑰红色产物
巴比妥和其他无芳环取代的巴比妥类药物无此反应---用于区别
特征熔点行为
巴比妥类药物的钠盐溶于水,酸化后析出游离巴比妥母体--沉淀--过滤--洗涤--干燥---测定熔点
也可以衍生化为衍生物后--测定衍生物的熔点
红外吸收光谱
中国药典2015--几乎所有的巴比妥类药物的原料药都采用此法作为鉴别方法
标准图谱对照法
特殊杂质检查
苯巴比妥的特殊杂质检查
酸度
主要是控制副产物苯基丙二酰脲,产生原因是乙酰化不完全,
苯基丙二酰脲的酸性比苯巴比妥强,能使甲基橙指示剂呈红色
甲基橙的变色范围--3.2--4.4 红色--橙色-黄
取本品--加水--煮沸--放冷--滤过--滤液--甲基橙--不显红色
乙醇溶液的澄清度
主要控制苯巴比妥中间体Ⅰ的量
杂质在乙醇溶液中的溶解度比苯巴比妥小
取本品---乙醇--加热回流--溶液澄清
中性或碱性物质
控制中间体Ⅰ形成的酰胺,酰脲或分解产物等杂质
利用杂质和苯巴比妥在氢氧化钠试液和乙醚中的溶解性能不同
提取重量法
取本品--分液漏斗--氢氧化钠--水--乙醚--分层--乙醚层--水振荡洗涤3次--过滤--滤液--蒸干--滤渣不得过3mg
有关物质
HPLC
辛烷基硅烷键合硅胶
供试品溶液
对照溶液
不加校正因子的主成分自身对照法
司可巴比妥的特殊杂质检查
溶液的澄清度
控制水不溶性杂质
司可巴比妥钠和杂质在水溶性上的差异
本品--水--溶液澄清
中性或碱性物质
控制酰脲,酰胺类副产物
含量测定
银量法
与重金属离子反应,定量生成盐
溴量法
酸碱滴定法
紫外分光光度法
高效液相色谱法
苯并二氮杂卓类
第十一章 酚噻嗪类抗精神病药物的分析
硫氮杂蒽母核结构---三环共轭。2位连有电负性较大的基团,10位连有碱性侧链
性质
弱碱性---母环上的氮原子碱性很弱,但是10位侧链的氮原子具有很强的碱性
易氧化性
硫氮杂蒽母核上的硫具有还原性,可以被硫酸,硝酸,过氧化氢,三氯化铁等氧化显色
与金属离子配位显色
硫原子具有孤对电子,可以与金属钯离子配位显色
其氧化物没有此特性
紫外吸收
硫氮杂蒽母核共轭结构---取代基不同,出现不同程度红移,向波长长的方向移动。205,254,300nm
红外吸收
鉴别
一般选择2-4种不同原理的分析方法组成一组鉴别试验
化学法
与生物碱沉淀剂反应
碱性--生物碱沉淀剂反应
盐酸氯丙嗪
三硝基苯酚--沉淀,测熔点
盐酸氯丙嗪片
氧化显色反应
氧化剂--硫酸,硝酸,过氧化氢,三氯化铁,硫酸铈铵---显色
与钯离子配合显色反应
硫氮杂蒽母核的硫元素与钯离子反应--不受母核氧化物亚砜和砜的影响
含卤素取代基的反应
焰色反应
奋乃静的火焰显绿色
显色反应
氟取代基---有机破坏--酸性条件--茜素锆--红色变黄色
碳酸钠加碳酸钾--有机破环
小火加热蒸干排出溶剂对分析的干扰
氯化物的鉴别反应
酚噻嗪类盐酸盐药物
与硝酸银的沉淀反应
盐酸盐---硝酸与母核会发生氧化显色反应,影响观察,因此加入氨溶液,使碱性的酚噻嗪类药物沉淀后,在进行氯化物的沉淀检查
与二氧化锰等氧化剂的氧化还原反应
氯离子与二氧化锰等氧化剂的氧化还原反应用于鉴别酚噻嗪类药物的盐酸盐,会生成氯气,使 湿润的淀粉碘化钾试纸变蓝--母核与氧化剂的显色反应不会影响观察
光谱法
红外分光光度法
紫外分光光度法
避光操作,254,300nm处。
色谱法
薄层色谱法
氢氧化铵作为扫尾剂--抑制弱碱性奋乃静与硅胶酸性集团的结合,减轻色谱斑点的拖尾程度
HPLC法
色谱峰的保留时间一致
其他方法
测定熔点的方法
毛细管法--测定初熔--熔矩等
有关物质检查
有关物质
残留的原料和中间产物--副产物--药物的氧化产物
检查方法--HPLC,TLC
盐酸氯丙嗪及其制剂的有关物质检查
ChP
HPLC-不加校正因子的主成分自身对照法
EP
HPLC--杂质对照品法--不加校正因子的主成分自身对照法
USP
TLC法--自身稀释对照品法
奋乃静及其制剂的有关物质检查
奋乃静
系统适用性试验--过氧化氢--放置1.5h
奋乃静片
含量测定
第十三章
莨菪烷类抗胆碱药物的分析
主要理化性质
水解性
酯的水解--生成莨菪醇和莨菪酸
碱性
叔胺氮原子或者季胺氮原子,具有较强的碱性
旋光性
东莨菪碱--比旋度
阿托品没有旋光性---可以用该性质区别阿托品和东莨菪碱
鉴别实验
托烷生物碱的vitali鉴别反应
生物碱--水解生成莨菪酸--发烟硝酸加热--三硝基衍生物--氢氧化钾-乙醇-氢氧化钾固体--有色的醌型产物--深紫色
与硫酸-重铬酸钾的反应
本类药物--硫酸水解--生成莨菪酸--硫酸-重铬酸钾加热---苯甲醛/苦杏仁的臭味
与生物碱显色剂和沉淀剂的反应
生物碱药物--生物碱沉淀剂---沉淀
氯化汞醇,碘化铋钾,三硝基苯酚,硅钨酸等
光谱鉴别法
uv
通过比较最大波长,最小波长或吸收光谱的一致性进行鉴别,也可以比较其吸光度或吸收系数的比值来鉴别
IR
绝大多数生物碱原料药都采用ir法鉴别
色谱鉴别法
一般用于已知生物碱的鉴别
TLC--HPLC--GC--PC
硫酸盐和溴化物的反应
特殊杂质的检查
氢溴酸东莨菪碱
特殊杂质的检查
酸度
碱性很弱--强酸弱碱盐---5%水溶液ph值为4-5.5控制本品的酸性杂志
易氧化物
杂质含有不饱和双键---样品--高猛酸钾--10分钟内红色不得完全消失
紫外吸光度法检查
其他生物碱
加入氨试液--不应该浑浊-----加入氢氧化钾---只许发生瞬即消失的类白色浑浊
硫酸阿托品--有关物质的检查
莨菪碱---旋光度测定法
有关物质--HPLC法
含量测定
酸性染料比色法
生物碱性药物--ph--与氢离子给合成阳离子--与酸性染料结合--红移
影响因素
水相的最佳ph值
酸性染料及其浓度
与有机碱定量结合
生成的离子对在有机相中具有较大的溶解度
染料在有机相中不容或微溶
有机溶剂的选择
对离子对萃取效率高
不与或极少与水混溶
水分的影响
水相中有过量酸性染料
水分混入使三氯甲烷浑浊
脱水剂,干燥滤纸过滤
酸性染料中的有机杂质
先用有机溶剂萃取缓冲溶液与酸性染料的混合溶液,然后再加入药物
非水溶液滴定法
弱碱性--高氯酸冰醋酸中硫酸是一级解离
高氯酸,冰醋酸,醋酐,结晶紫
HPLC
反向高效--离子对
碱性药物
离子对试剂---烷基磺酸盐
酸性药物
季铵盐
第十五章 甾体激素类药物的分析
结构与分类
肾上腺皮质激素类
🔺4-3-酮-----UV,羰基试剂显色
C17-α-酮醇基-----还原性
卤素---元素反应
雄激素与蛋白同化激素
🔺4-3-酮----uv,羰基试剂显色
C17-β羟基---成酯
孕激素
🔺4-3-酮--uv,羰基试剂显色
C17-甲基酮--亚硝基铁氰化钠
C17-乙炔基---硝酸银反应
雌激素
A环为苯环,UV,重氮苯磺酸盐反应
C17-乙炔基--硝酸银反应
C17-羟基---成酯
理化性质与鉴别试验
性状特征
性状与溶解度
熔点
比旋度
吸收系数
化学鉴别法
与强酸的显色反应
酮基质子化--碳正离子--HSO4反应--显色
官能团的反应
C17-α醇酮基的呈色反应
具有还原性,与羰基试剂--四氮唑,多伦,斐林试剂--显色。四氮唑试剂--甲囋显色
酮基的显色反应---C3.C20;--2,4-二硝基本肼,硫酸苯肼,异烟肼--黄色的腙
C17-甲基酮的呈色反应---黄体酮的专属,灵敏的鉴别方法
与亚硝基铁氰化钠反应---蓝紫色
酚羟基的呈色反应
重氮苯磺酸--红色偶氮染料
炔基的沉淀反应
硝酸银反应--白色炔银沉淀
卤素的反应
氟原子
茜素氟蓝硝酸亚铈--蓝紫色
氯原子
先破坏--在反应
酯的反应
一般先水解,生成相应的羧酸--在鉴别
醋酸酯类--在硫酸存在条件下与乙醇形成乙酸乙酯,香气鉴别
紫外-可见分光光度法
红外分光光度法
色谱法
HPLC法



Collect
Get Started

Collect
Get Started

Collect
Get Started

Collect
Get Started
评论
0 条评论
下一页

