2023版GMP无菌制剂——配制
2024-04-11 15:42:35 2 举报AI智能生成
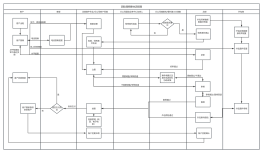
根据2023版GMP规范,无菌制剂的配制过程涉及到关键质量属性(CQAs)的确定、原材料的选择和控制、生产过程的设计和优化、生产环境的控制、人员培训和资格认证、过程验证和持续改进等多个方面。其中,思维的引入和加强是实现产品安全生产和满足患者需求的关键。它要求我们从产品设计和生产过程之初就引入风险评估和预防思维,充分考虑患者的需求,同时从质量、效率和成本的角度进行全面系统的设计。此外,在整个生产过程中,应保持思维的持续性和灵活性,根据实际情况进行动态调整和优化,以确保产品的质量和安全。因此,2023版GMP无菌制剂思维配制是一种以患者需求为中心,以质量为基石,以风险管理和持续改进为驱动的配制理念和方法。
模板推荐
作者其他创作
大纲/内容








0 条评论
下一页

