医疗器械开发注册流程
2025-03-26 11:31:23 11 举报AI智能生成
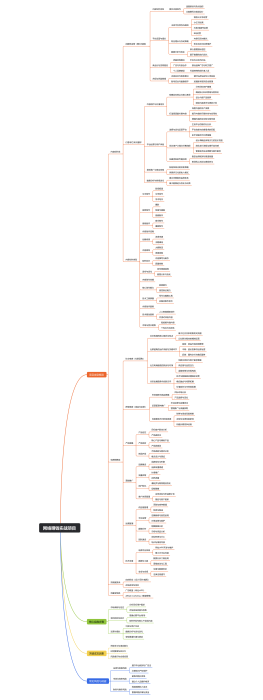
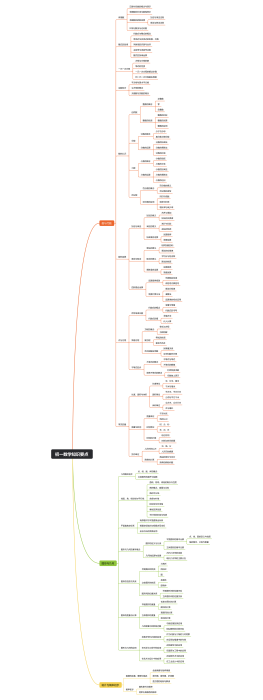
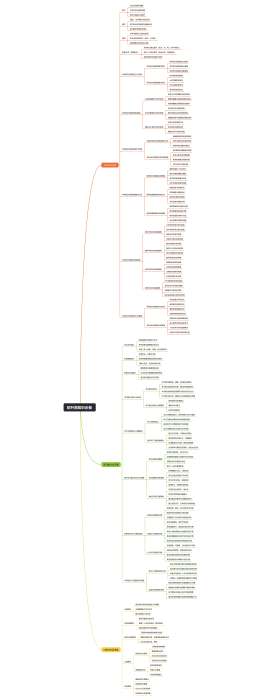
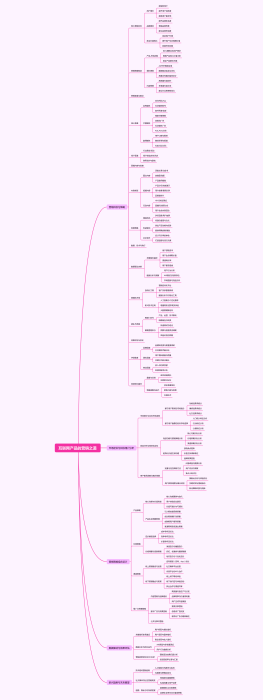
医疗器械开发注册流程是一系列步骤,确保产品从概念设计到市场销售的安全性、有效性和合规性。首先,在研发阶段,需进行产品设计和风险分析,编写技术文档,并建立临床前测试。接着,在注册阶段,企业需准备和提交注册文件,其中包含产品详细说明和临床评估报告,以满足监管机构(如美国食品药品监督管理局FDA或欧盟的欧洲药品管理局EMA)的要求。此外,可能会进行临床试验以支持产品的安全性和有效性。 注册文档通常须包括详细的产品资料,如结构、功能和性能描述;必须符合适当的医疗器械法规,例如医疗器械指令(MDD)或医疗器械法规(MDR);和预期用途。此流程要求精确、详实的报告,并严格遵守制药法规及行业标准。过程中,文件和数据的组织性、完整性和可追踪性是成功注册的决定性因素。修饰语应用于强调严格性和精准度,可能包括“详尽严格的”、“确保合规性”或“彻底验证的”等。最终目的是为了患者健康和安全,确保医疗产品达到所需的质量标准和监管要求。
模板推荐
作者其他创作
大纲/内容




0 条评论
下一页

