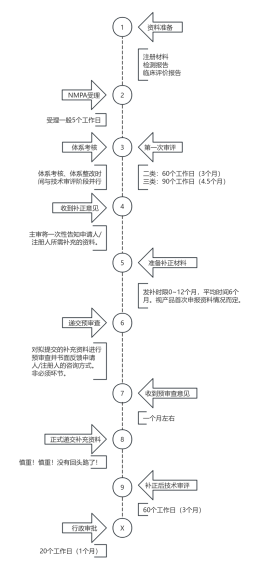
4.1一般文件
4.1.1填妥的中成药注册申请专用表格;
4.1.2申请人的识别数据及相关证明文件、刑事纪录证明书及未有任何债务正透过税务执行程序进行强制征收的证明文件,相关文件须符合第46/2021号行政法规《中药药事活动及中成药注册法施行细则》第六十五条第二款(一)项(1)分项至(3)分项的规定;
4.1.3倘中成药注册申请属中药制药厂或研发者作出,申请人须声明其有权提出中成药注册申请;倘属进口中成药注册申请,申请人须提供由原产国或地区,又或来源国或地区主管当局认可的中成药持有人或制造商发出的可作中成药注册申请的授权书;
4.1.4包装、标签及倘有的说明书的样本或式样。在取得中成药注册后,注册持有人须提交完整的中成药包装、标签及倘有的说明书的实物样本;
4.1.5提供由中药制药厂或具资质的检验机构,包括但不限于内地市级或以上的药品检验机构、或已取得ISO17025、中国计量认证(CMA)或中国合格评定国家认可委员会(CNAS)认证相关检验资质的检验机构发出的样品检验报告,而所送交的样品必须由申报注册所述的制造商厂址内制造;
4.1.6原产国或地区发出的制造商生产许可的官方文件正本或经认证的副本,如属药物监督管理局发出准照的中药制药厂可无需递交;
4.1.7如属委托制造中成药的情况,相关委托的证明文件,尤其是委托许可,又或委托合同的拟本或该合同的副本,如属药物监督管理局许可的委托制造可无需递交;
4.1.8倘有的原产国或地区、又或出口国或地区主管当局发出的中成药注册或销售证明书正本或经认证的副本;
4.1.9已遵守《濒危野生动植物种国际贸易公约》的证明文件,尤指经济及科技发展局发出的濒危野生动植物种国际贸易公约(CITES)的证明书副本,又或其他国家或地区主管当局发出的等同文件,不含濒危野生动植物种除外;
4.1.10如申请人属申请注册中成药的专利持有人或其委托人,须提交持有专利保护的声明及证明文件。上述声明须载有中成药专利的卷宗数据,尤其是专利的标的、所赋予的权利及保护期间;如申请人属非申请注册中成药的专利持有人或其委托人,则须提交表明其不会亦无意违反有关专利保护的声明;
4.1.11倘有的载有以数据资料的保护为目的而应予保密的数据及其保密理由的文件。根据第11/2021号法律《中药药事活动及中成药注册法》第三十三条的规定,相关数据资料为药理学、毒理学研究及临床试验的数据。
4.2药学研究资料
4.2.1药学研究总结报告
对中药<b><font color="#e74f4c">成分研究</font></b>、<b><font color="#e74f4c">剂型选择</font></b>、<b><font color="#e74f4c">工艺研究</font></b>、<b><font color="#e74f4c">质量控制研究</font></b>及<b><font color="#e74f4c">稳定性研究</font></b>的结果进行总结,综合分析及评价中成药<b><font color="#4669ea">质量控制情况</font></b>。
4.2.2各中药成分的来源及质量控制
4.2.2.1中药材或中药饮片的来源
包括<b><font color="#e74f4c">基原</font></b>,须注明<b><font color="#e74f4c">科名</font></b>、<b><font color="#e74f4c">中文名</font></b>及<b><font color="#e74f4c">拉丁学名</font></b>,以及<b><font color="#e74f4c">药用部位</font></b>,如属<b><font color="#4669ea">矿物药</font></b>须注明<b><font color="#e74f4c">类</font></b>、<b><font color="#e74f4c">族</font></b>、<b><font color="#e74f4c">矿石名</font></b>或<b><font color="#e74f4c">岩石名</font></b>、<b><font color="#e74f4c">主要成分</font></b>。
4.2.2.2中药材或中药饮片的质量资料
4.2.2.2.1中药材或中药饮片的<b><font color="#e74f4c">执行标准</font></b>或<b><font color="#4669ea">自拟</font></b>的<b><font color="#e74f4c">质量标准</font></b>;
4.2.2.2.2中药材或中药饮片的<b><font color="#e74f4c">检验报告</font></b>;
4.2.2.2.3倘为<b><font color="#4669ea">新药材</font></b>,须提供药材<b><font color="#e74f4c">生态环境</font></b>、<b><font color="#e74f4c">形态描述</font></b>、<b><font color="#e74f4c">生长特征</font></b>、<b><font color="#e74f4c">种植或养殖技术</font></b>,以及相关的<b><font color="#e74f4c">标本</font></b>。
4.2.2.3中药饮片的炮制工艺,须提供中药饮片<b><font color="#e74f4c">炮制工艺依据</font></b>及<b><font color="#e74f4c">详细工艺参数</font></b>;
4.2.2.4中药材或中药饮片的供应商数据,属<b><font color="#e74f4c">新药材</font></b>及被载于<b><font color="#4669ea">《澳门特别行政区所用中药材表》</font></b>的<b><font color="#e74f4c">毒性中药材及其饮片</font></b>尚须提供其来源单据;
4.2.2.5倘属<b><font color="#e74f4c">外购中药提取物</font></b>,须提供相关中药提取物的<b><font color="#e74f4c">质量标准</font></b>、<b><font color="#e74f4c">制备方法</font></b>、<b><font color="#e74f4c">制造商数据</font></b>,以及倘有的供应商数据及<b><font color="#e74f4c">批准文件</font></b>;
4.2.2.6倘属<b><font color="#e74f4c">自制提取物</font></b>,须提供所用<b><font color="#e74f4c">饮片的相关数据</font></b>、<b><font color="#e74f4c">详细的制备工艺</font></b>及<b><font color="#e74f4c">工艺研究数据</font></b>;
4.2.2.7倘中成药含<b><font color="#e74f4c">牛源性原料</font></b>,必须同时符合<b><font color="#4669ea">《牛源性中成药注册技术性规范》</font></b>的技术性指示之要求。
4.2.3配制方法
4.2.3.1中成药配方,提供<b><font color="#e74f4c">1000个制剂单位</font></b>的各中药成分及辅料的配方组成资料;
4.2.3.2制备工艺
4.2.3.2.1工艺研究资料
提供制备<b><font color="#e74f4c">工艺路线</font></b>筛选研究资料,说明制备工艺路线选择的<b><font color="#e74f4c">合理性</font></b>。
如中成药配方来源于<b><font color="#e74f4c">医院制剂</font></b>、<b><font color="#e74f4c">临床验方</font></b>或具<b><font color="#e74f4c">有人用经验</font></b>的,应详细说明在临床应用时的<b><font color="#4669ea">具体使用情况</font></b>;
属<b><font color="#4669ea">改良型新药</font></b>须说明与<b><font color="#4669ea">原制剂生产工艺</font></b>的<b><font color="#e74f4c">异同</font></b>及<b><font color="#e74f4c">参数的变化情况</font></b>;
4.2.3.2.2详细描述制备工艺
对制备工艺的过程进行规范描述,明确工艺流程及工艺参数;
4.2.3.2.3工艺流程图
按制备工艺步骤提供完整、直观、简洁的工艺流程图,应涵盖所有的工艺步骤,标明主要工艺参数和所用提取溶剂;
4.2.3.2.4中药成分、辅料及溶剂的含量清单,对于制剂工艺中使用但最后去除的溶剂也应列出;
4.2.3.2.5说明<b><font color="#4669ea">剂型选择</font></b>及<b><font color="#4669ea">规格确定</font></b>的<b><font color="#e74f4c">依据</font></b>;
4.2.3.2.6主要生产设备数据;
4.2.3.2.7倘申请注册的中成药已获原产国或地区,又或来源国或地区主管当局核准注册,可仅提供第4.2.3.2.2点至第4.2.3.2.4点所指的数据。
4.2.4辅料的质量控制
所用的辅料应具有标准,并提供<b><font color="#e74f4c">辅料的来源和执行标准</font></b>。
4.2.5中成药的质量标准
4.2.5.1质量研究的试验数据及文献资料
提供与质量标准建立相关的质量研究数据,包括但不限于反映中成药<b><font color="#e74f4c">关键质量的检测指标</font></b>选择及<b>质控方法</b>,并提供相关文献资料;
4.2.5.2质量标准及起草说明,相关质量标准须符合或<b><font color="#e74f4c">不低于</font></b>已有的<b><font color="#4669ea">药典</font></b>或<b><font color="#4669ea">标准</font></b>的要求;
4.2.5.3制成品质量检验报告
4.2.5.3.1提交最少<b><font color="#4669ea">三个</font><font color="#e74f4c">最新批次</font></b>的制成品质量检验报告;
4.2.5.3.2提交最少三个最新批次的制成品符合<b><font color="#4669ea">《中成药重金属及有毒元素含量、微生物限度和农药残留限量标准规范》</font></b>的检验报告;
4.2.5.3.3如属<b><font color="#4669ea">改良型新药</font></b>或<b><font color="#4669ea">创新药</font></b>,则须提交具资质的检验机构发出的<b><font color="#e74f4c">质量标准复核报告</font></b>。
4.2.5.4倘申请注册的中成药已获原产国或地区,又或来源国或地区主管当局核准注册,可提供原产国或地区,又或来源国或地区主管当局所核准的质量标准替代第4.2.5.1点及第4.2.5.2点所指的数据。
4.2.6稳定性
4.2.6.1稳定性试验
4.2.6.1.1稳定性试验研究资料的内容至少包括试验方法、检测项目、试验时间、结果分析、有效期的确定、储存条件的确定、相关检测图谱;
4.2.6.1.2稳定性总结报告
须总结稳定性研究的样品情况、考察条件、考察指标和考察结果,并拟定储存条件及有效期;
4.2.6.1.3获注册后的稳定性研究方案及声明;
4.2.6.1.4倘申请注册的中成药已获原产国或地区,又或来源国或地区主管当局核准注册,可仅提供第4.2.6.1.2点所指的数据。
4.2.6.2直接接触中成药的包装材料和容器的选择
声明所采用的包装材料符合质量标准,并提供倘有的包装材料和容器的质量标准和合法来源的证明文件。
4.2.7倘有的药学研究参考文献。
4.3药理毒理学研究资料
药理毒理学研究须按照人用药品注册技术要求国际协调会(ICH)建议,并在符合《药物非临床研究质量管理规范》(GLP)的机构开展,遵守GLP规范。
4.3.1药理毒理学研究总结报告
药理毒理学研究总结报告是对药理学、药代动力学、毒理学研究的综合性和关键性评价。
4.3.1.1药理毒理学试验策略概述
提供试验样品的制备工艺,结合申请的注册类别、处方来源或人用经验数据、所申请的功能主治等,介绍药理毒理学试验的研究思路及策略;
4.3.1.2药理学研究总结
简要概括药理学研究内容,按概要、主要药效学、次要药效学、安全药理学、药效学药物相互作用、讨论及结论的顺序撰写,并附列表总结;
4.3.1.3药代动力学研究总结
简要概括药代动力学研究内容,按概要、分析方法、吸收、分布、代谢、排泄、药代动力学药物相互作用、其他药代动力学试验、讨论及结论的顺序撰写,并附列表总结;
4.3.1.4毒理学研究总结
简要概括毒理学试验结果,并说明试验的GLP 依从性,说明毒理学试验受试物情况。
按概要、单次给药毒性试验、重复给药毒性试验、遗传毒性试验、致癌性试验、生殖毒性试验、制剂安全性试验(刺激性、溶血性、过敏性试验等)、其他毒性试验、讨论和结论的顺序撰写,并附列表总结;
4.3.1.5综合分析与评价
4.3.1.5.1对药理学、药代动力学、毒理学研究进行综合分析与评价;
4.3.1.5.2分析药理学、药代动力学与毒理学结果之间的相关性;
4.3.1.5.3结合药学、临床数据进行综合分析与评价。
4.3.1.6参考文献
提供有关的参考文献,必要时应提供全文。
4.3.2药理学研究资料
药理学研究是通过动物或体外、离体试验来获得非临床有效性信息,包括药效学作用及其特点、药物作用机制等。
4.3.2.1主要药效学
4.3.2.2次要药效学
4.3.2.3安全药理学
4.3.2.4药效学药物相互作用
4.3.3药代动力学研究资料
非临床药代动力学研究是通过体外和动物体内的研究方法,揭示药物在体内的动态变化规律,获得药物的基本药代动力学参数,阐明药物的吸收、分布、代谢和排泄的过程和特征。
4.3.3.1分析方法及验证报告
4.3.3.2吸收
4.3.3.3分布
4.3.3.4代谢
4.3.3.5排泄
4.3.3.6药代动力学药物相互作用(非临床)
4.3.4毒理学研究数据
4.3.4.1单次给药毒性试验
4.3.4.2重复给药毒性试验
4.3.4.3遗传毒性试验
4.3.4.4生殖毒性试验
4.3.4.5致癌性试验
4.3.4.6依赖性试验
4.3.4.7刺激性、过敏性、溶血性等与局部、全身给药相关的制剂安全性试验
4.3.5第4.3.2点至4.3.4点所指的试验,其报告的内容须包括试验方法、结果及总结等有关资料
4.4临床研究资料
临床研究须按照人用药品注册技术要求国际协调会(ICH)建议,并在符合《药物临床研究质量管理规范》(GCP)的机构开展,遵守GCP规范。
4.4.1临床研究总结
4.4.1.1根据注册分类提供相应的简要中医药理论、或人用经验对拟定功能主治的支持情况评价,拟选择调节病症的病因、病机、治疗等;
4.4.1.2人用经验概况<br>
如有人用经验的,须提供简要人用经验概述,并分析说明人用经验对拟定功能主治或后续所需开展临床试验的支持情况;
4.4.1.3分析与评价<br>
从立题目的、立题依据、临床试验方案的合理性和可行性对申请注册的中成药进行客观的综合分析与评价。
以临床试验结果为依据,对试验中成药安全性进行分析及评价,分析受试中成药的可能的高风险人群。
阐述安全性问题对受试中成药临床广泛应用的可能影响。
根据分析与评价结果,归纳功能主治或适应症、用法用量、不良反应、禁忌、注意事项等内容;
4.4.1.4参考文献
提供有关的参考文献,必要时应提供全文。
4.4.2立题目的与依据
4.4.3临床试验研究资料
4.4.3.1临床试验方案
4.4.3.2由进行临床试验当地的主管当局发出的批准进行临床试验的证明文件副本,如属药物监督管理局许可的临床试验除外;
4.4.3.3知情同意书式样
4.4.3.4研究者手册
4.4.3.5临床试验报告及其附件。包括但不限于:
4.4.3.5.1临床试验报告
4.4.3.5.2病例报告表式样、患者日志
4.4.3.5.3临床试验主要有效性、安全性数据相关的关键标准操作规程
4.4.3.5.4临床试验方案变更情况说明
4.4.3.5.5统计分析计划及报告
4.4.4人用经验的证明性文件
4.4.5既往临床应用情况概述及总结报告